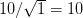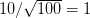Well, it certainly has been a while since I've written anything here. Life has gotten busy with new projects, new responsibilities, etc. Yesterday, I participated in a workshop on campus sponsored by the Woods Institute for the Environment, the Young Environmental Scholars Conference. I was asked to stand-in for a faculty member who had to cancel at the last minute. I threw together some rather hastily-written notes and figured I'd share them here (especially since I spoke quite a bit of the importance for public communication!).
The theme of the conference was "Environmental Policy, Behavior, and Norms" and we were asked to answer three questions: (1) What does doing normative research mean to you? (2) How do your own norms and values influence your research? (3) What room and role do you see for normative research in your field? So, in order, here are my answers.
What does doing normative research mean to you?
I actually don't particularly like the term "normative research" because it sounds a little too much like imposing one's values on other people. I am skeptical of the imposition of norms that have more to do with (often unrecognized) ideology and less about empirical truth – an idea that was later reinforced by a terrific concluding talk by Debra Satz. If I can define "normative" to mean with the intent to improve people’s lives, then OK. Otherwise, I prefer to do "positive" research.
For me, normative research is about doing good science. As a biosocial scientist with broad interests, I wear a lot of hats. I have always been interested in questions about the natural world, and (deep) human history in particular. However, I find that the types of questions that really hold my interest these days are more and more engaged in the substantial challenges we face in the world with inequality and sustainability. In keeping with my deep pragmatist sympathies, I increasingly identify with Charles Sanders Pierce's idea that given the "great ocean of truth" that can potentially be uncovered by science, there is a moral burden to do things that have social value. (As an aside, I think that there is social value in understanding the natural world, so I don’t mean to imply a crude instrumentalism here.) In effect, there is a lot of cool science to be done; one may as well do something of relevance. I personally have little patience for people who pursue racist or otherwise socially divisive agendas and cloak their work in a veil of free scientific inquiry. This said, I worry when advocacy interferes with intellectual fairness or an unwillingness to accept that one's position is not actually true.
I think that we are fooling ourselves if we believe that our norms somehow don't have an effect on our research. Recognizing what these norms that shape your research – whether implicitly or explicitly – helps you manage your bias. Yes, I said manage. I'm not sure we can ever completely eliminate it. I see this as more of a management of a necessary trade-off, drawing an analogy between the practice of science and a classic problem in statistics, between bias and variance. The more biased one is, the less variance there is in the outcome of one’s investigation. The less bias, the greater the likelihood that results will differ from one’s expectations (or wishes). Recognizing how norms shape our research also deals with that murky area of pre-science: where do our ideas for what to study come from?
How do your own norms and values influence your research?
Some of the the norms that shape my own research and teaching include:
transparency: science works best when it is open. This places a premium on sharing data, methods, and communicating results in a manner that maximizes access to information. As a simple example, this norm shapes my belief that we should not train students from poor countries in the use of proprietary software (and other technologies) that they won't be able to afford when they return to their home countries when there are free or otherwise open-source alternatives.
fairness: this naturally includes a sense of social justice or people playing on an equal playing field, but it also includes fairness to different ideas, alternative hypotheses, the possibility that one is wrong. This type of fairness is essential for one's credibility as a public intellectual in science (particularly supporting policy), as noted eloquently in this interview with Dick Lewontin.
respect for people's ultimate rationality: Trying to understand the social, ecological, and economic context of people's decision-making, even if it violates our own normative – particularly market-based economic – expectations.
flexibility: solving real problems means that we need to be flexible in our approach, willing to go where the solutions lead us, learning new tools and collaborating. Flexibility also means a willingness to give up on a research program that is doing harm.
good-faith communication: I believe that there is no room for obscurantism in the academy of the 21st century. This includes public communication. There are, of course, complexities here with regard to the professional development of young scholars. One of the key trade-offs for young scholars is the need for professional advancement (which comes from academic production) and activism, policy, and public communication. Within the elite universities, the reality is that neither public communication nor activism count much for tenure. However, as Jon Krosnick noted, tenure is a remarkable privilege and, while it may seem impossibly far away for a student just finishing a Ph.D., it’s not really. Once you prove that you have the requisite disciplinary chops, you have plenty of time to to use tenure for what it is designed for (i.e., protecting intellectual freedom) and engaging in critical public debate and communication.
humility: solving problems (in science and society) means caring more about the answer to a problem than one's own pet theory. Humility is intimately related to respect for others' rationality. It also means recognizing the inherently collaborative nature of contemporary science: giving credit where it is due, seeking help when one is in over one’s head, etc. John DeGioia, President of Georgetown University, quoted St. Augustine in his letter of support for Georgetown Law Student, Sandra Fluke against the crude attacks by radio personality Rush Limbaugh and I think those words are quite applicable here as well. Augustine implored his interlocutors to "lay aside arrogance" and to "let neither of us assert that he has found the truth; let us seek it as if it were unknown to both." This is not a bad description of the way that science really should work.
What room and role do you see for normative research in your field?
I believe that there is actually an enormous amount of room for normative research, if by "normative research," we mean research that has the potential to have a positive effect on people's lives. If instead we mean imposing values on people, then I am less sure of its role.
Anthropology is often criticized from outside the field, and to a lesser extent, from within it for being overly politicized. You can see this in Nicholas Wade’s critical pieces in the New York Times Science Times section following the American Anthropological Association’s executive committee excising of the word "science" from the field’s long-range planning document. Wade writes,
The decision [to remove the word 'science' from the long-range planning document] has reopened a long-simmering tension between researchers in science-based anthropological disciplines — including archaeologists, physical anthropologists and some cultural anthropologists — and members of the profession who study race, ethnicity and gender and see themselves as advocates for native peoples or human rights.
This is a common sentiment. And it is a complete misunderstanding. It suggests that scientists can't be advocates for native peoples or human rights. It also suggests that one can't study race, ethnicity, or gender from a scientific perspective. Both these ideas are complete nonsense. For all the leftist rhetoric, I am not impressed with the actual political practice of what I see in contemporary anthropology. There is plenty of posturing about power asymmetries and identity politics but it is always done in such a mind-numbingly opaque language and with no apparent practical tie-in to policies that make people's lives better. And, of course, there is the outright disdain for "applied" work one sees in elite anthropology departments.
Writing specifically about Foucault, Chomsky captured my take on this whole mode of intellectual production:
The only way to understand [the mode of scholarship] is if you are a graduate student or you are attending a university and have been trained in this particular style of discourse. That's a way of guaranteeing...that intellectuals will have power, prestige and influence. If something can be said simply, say it simply, so that the carpenter next door can understand you. Anything that is at all well understood about human affairs is pretty simple.
Ultimately, the simple truths about human affairs that I find anyone can relate to are subsistence, health, and the well-being of one’s children. These are the themes at the core of my own research and I hope that the work I do ultimately can effect some good in these areas.